Desmond_2022-4在HPC上的表现和批量化MD操作
拳打Amber脚踢Gromacs最终攀上Desmond高峰
一、说明:
笔者在长期受到Amber、Gromacs模拟系统构建的折磨后,痛定思痛决定下载带有GUI美观、完全自动化、一键分析的Desmond。上海科技大学Wang-Lin搭建了Schrödinger中文社区微信群(公众号:蛋白矿工),实现了使用者的互相交流和问答摘录整理,并就Schrödinger软件的使用编写了大量实用脚本。
基于命令行操作的Amber代码包非常适合生物分子体系的模拟,包括AmberTools、Amber两部分。前者免费,但提升生产力的多CPU并行和GPU加速功能需要通过Amber实现,但发展中国家(如中国)可以通过学术申请获得。
同样基于命令行操作的的Gromacs代码包由于上手快、中文教程详尽等优点收到初学者的一致好评,教程可以参考李继存老师博客(http://jerkwin.github.io/)、B站和计算化学公社(http://bbs.keinsci.com/forum.php)等。 初学者跟着教程做就可以(http://www.mdtutorials.com/),深入学习还是推荐大家去看官方的文档。
初识Desmond是使用世界上最贵的分子模拟软件之一的“学术版”Maestro时,它是由D.E.Shaw Research开发的MD代码,但在Schrödinger公司的帮助下实现了GUI图形界面操作。投资界科学家D.E.Shaw顺手还制造l一台用于分子动力学模拟的专业超级计算机Anton2,当然相对于宇宙最快的Gromacs还是差了点(懂得都懂)。学术和非营利机构用户更是可以在D.E.Shaw Research官方网站注册信息,免费获得拥有Maestro界面的Desmond软件。
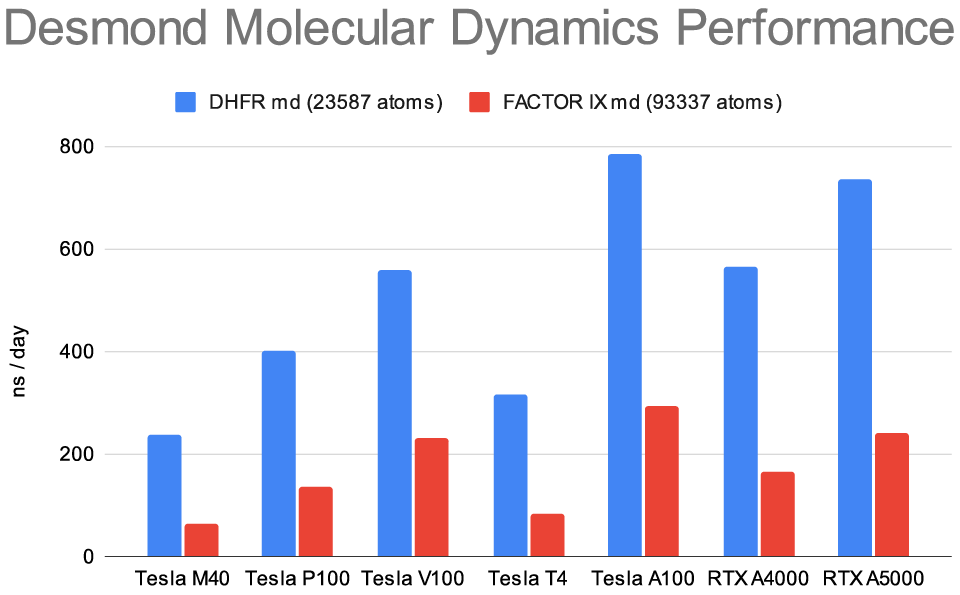
Figure 1: Molecular Dynamics run using default parameters in Schrödinger Suite 2023-1.
二、Desmonds的安装:
在D.E.Shaw Research注册后下载相应软件包,上传到HPC集群个人home下任意位置,随后创建软件文件夹(作为后续安装路径)复制pwd输出的路径内容(笔者为/fsa/home/ljx_zhangzw/software):
mkdir software
pwd解压安装,进入安装引导界面并按Enter继续进行安装:
tar -xvf Desmond_Maestro_2022.4.tar
cd DESRES_Academic_2022-4_Linux-x86_64
./INSTALL
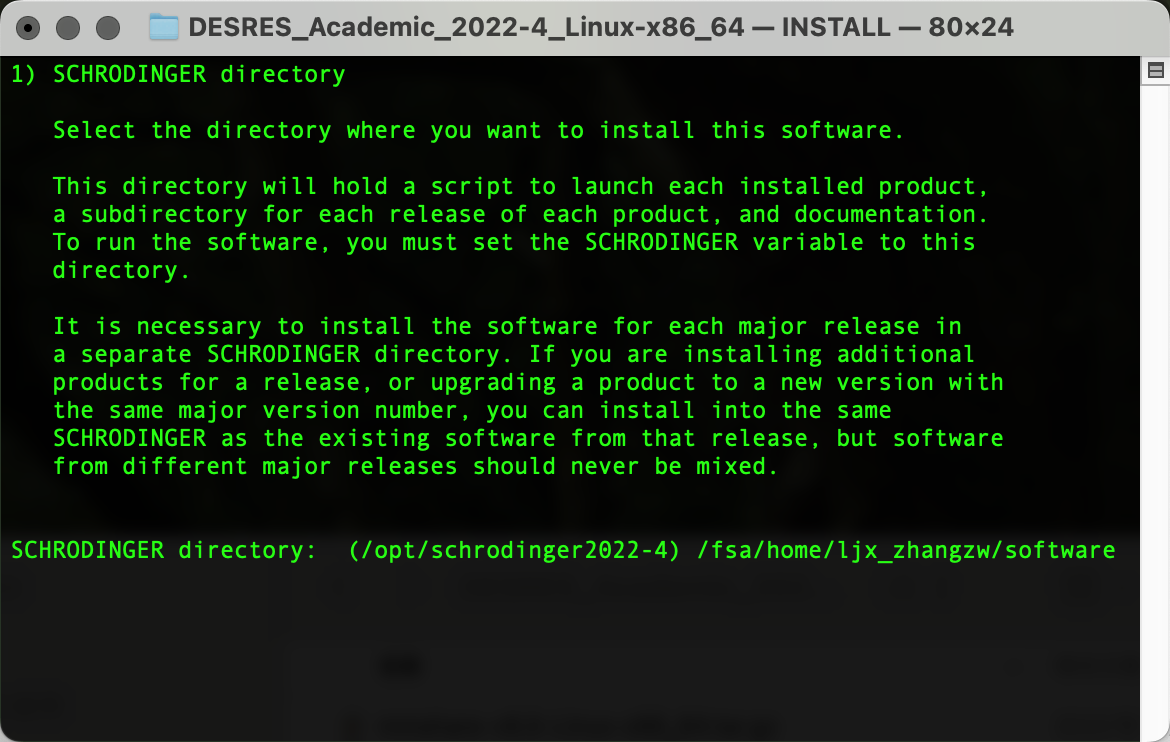
输出以下界面后粘贴复制pwd输出的路径内容,Enter确认为安装路径:
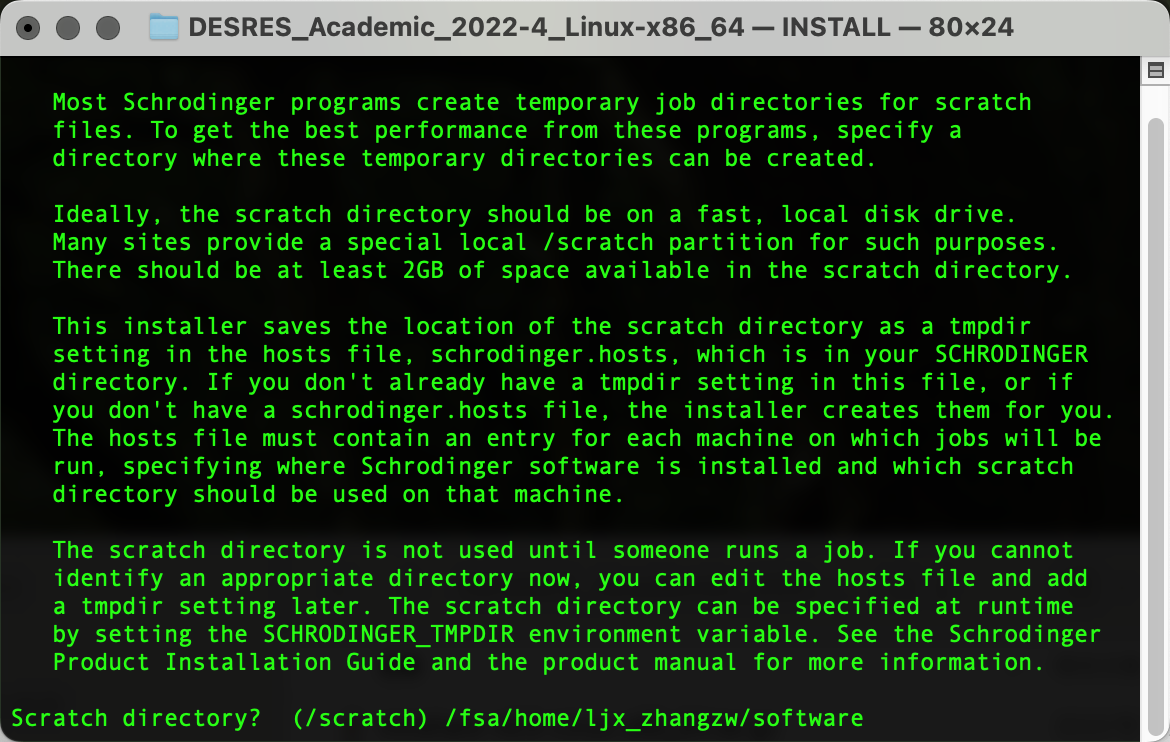
输出以下界面后粘贴复制pwd输出的路径内容,Enter确认为临时文件路径:
输出以下界面后输入y,Enter确认上述两个路径并进行安装:
安装完成后进入安装路径,并写入环境变量:
cd /fsa/home/ljx_zhangzw/software
echo "export Desmond=${PWD}/" >> ~/.bashrc同时由于Desmond对集群计算中提交任务队列的要求,需要在安装路径修改schrodinger.host以定义GPU信息,示例给出的是83a100ib队列中m001节点信息:
#localhost
name:localhost
temdir:/fsa/home/ljx_zhangzw/software
#HPC
name: HPC
host: m001
queue: LSF
qargs: -q 83a100ib -gpu num=1
tmpdir: /fsa/home/ljx_zhangzw/software
schrodinger: /fsa/home/ljx_zhangzw/software
gpgpu: 0, NVIDIA A100
gpgpu: 1, NVIDIA A100
gpgpu: 2, NVIDIA A100
gpgpu: 3, NVIDIA A100
gpgpu: 4, NVIDIA A100
gpgpu: 5, NVIDIA A100
gpgpu: 6, NVIDIA A100
gpgpu: 7, NVIDIA A100需要注意的是:
1、修改tmpdir和schrodinger对应路径为自己的安装路径;
2、节点host、队列83a100ib以及显卡信息gpgpu: 0-7, NVIDIA A100自行调整,可以查看HPC的计算资源;
3、对于不同的任务调度系统,Schrödinger公司KNOWLEDGE BASE和蛋白矿工知乎号Q2进行了介绍;
4、截止2023年6月16日,HPC集群可供Desmond使用的计算卡(而非3090、4090等)包括:
| 队列 | GPU | Hostname | 加速卡机时收费 | ||
| e5v3k40ib | 2*Intel Xeon E5-2680v3 2*NVIDIA Tesla K40 12GB 128GB RAM 56Gb FDR InfiniBand |
x001 gpgpu: 0, NVIDIA Tesla K40 gpgpu: 1, NVIDIA Tesla K40 |
校内及协同创新中心用户 1.2 元/卡/小时=0.1元/核/小时 |
1.2 | =0.1 |
| e5v4p100ib | 2*Intel Xeon E5-2660v4 2*NVIDIA Tesla P100 PCIe 16GB 128GB RAM 56Gb FDR InfiniBand |
x002 gpgpu: 0, NVIDIA Tesla P100 gpgpu: 1, NVIDIA Tesla P100 |
校内及协同创新中心用户 1.68 元/卡/小时=0.12元/核/小时 |
||
| 62v100ib | 2*Intel Xeon Gold 6248 8*NVIDIA Tesla V100 SXM2 32GB 768GB RAM 100Gb EDR InfiniBand |
n002 gpgpu: 0, NVIDIA Tesla V100 |
校内及协同创新中心用户 3 元/卡/小时=0.6元/核/小时 |
||
| 72rtxib | AMD EPYC 7302 4*NVIDIA TITAN RTX 24GB 128GB RAM 100Gb HDR100 InfiniBand |
g005 or g006 or g007 gpgpu: 0, NVIDIA TITAN RTX |
校内及协同创新中心用户 1.8 元/卡/小时=0.45元/核/小时 |
||
| 83a100ib | 2*Intel Xeon Platinum 8358 8*NVIDIA Tesla A100 SXM4 40GB 512GB RAM 200Gb HDR InfiniBand |
m001 gpgpu: 0, NVIDIA A100 |
校内及协同创新中心用户 4.8 元/卡/小时=0.6元/核/小时 |
三、运行MD模拟
单次MD:
在个人笔记本(Linux)上依照上述步骤安装,或使用Windows浏览器以图形界面Web登陆HPC集群进行点击操作。
教程可参考2022薛定谔中文网络培训和知乎内容。
批量MD:
1、plmd/DSMDrun: Desmond分子动力学模拟一键式运行脚本:
通过以上脚本实现当前路径下所有的mae文件(类似于PDB的结构文件,需通过Maestro转存)按照设置参数进行模拟,plmd -h中给出了示例和详细的输入命令介绍:
Usage: plmd [OPTION] <parameter>
An automatic Desmond MD pipline for protein-ligand complex MD simulation.
Example:
1) plmd -i "*.mae" -S INC -P "chain.name A" -L "res.ptype UNK" -H HPC_CPU -G HPC_GPU
2) plmd -i "*.mae" -S OUC -P "chain.name A" -L "chain.name B" -t 200 -H HPC_CPU -G HPC_gpu01
3) plmd -i "*.mae" -S "TIP4P:Cl:0.15-Na-Cl+0.02-Fe2-Cl+0.02-Mg2-Cl" -L "res.num 999" -G HPC_gpu03
4) plmd -i "*.cms" -P "chain.name A" -L "res.ptype ADP" -H HPC_CPU -G HPC_gpu04
Input parameter:
-i Use a file name (Multiple files are wrapped in "", and split by ' ') *.mae or *.cms ;
or regular expression to represent your input file, default is *.mae.
System Builder parameter:
-S System Build Mode: <INC>
INC: System in cell, salt buffer is 0.15M KCl, water is TIP3P. Add K to neutralize system.
OUC: System out of cell, salt buffer is 0.15M NaCl, water is TIP3P. Add Na to neutralize system.
Custom Instruct: Such as: "TIP4P:Cl:0.15-Na-Cl+0.02-Fe2-Cl+0.02-Mg2-Cl"
Interactive addition of salt. Add Cl to neutralize system.
for positive_ion: Na, Li, K, Rb, Cs, Fe2, Fe3, Mg2, Ca2, Zn2 are predefined.
for nagative_ion: F, Cl, Br, I are predefined.
for water: SPC, TIP3P, TIP4P, TIP5P, DMSO, METHANOL are predefined.
-b Define a boxshape for your systems. <cubic>
box types: dodecahedron_hexagon, cubic, orthorhombic, triclinic
-s Define a boxsize for your systems. <15.0>
for dodecahedron_hexagon and cubic, defulat is 15.0;
for orthorhombic or triclinic box, defulat is [15.0 15.0 15.0];
If you want use Orthorhombic or Triclinic box, your parameter should be like "15.0 15.0 15.0"
-R Redistribute the mass of heavy atoms to bonded hydrogen atoms to slow-down high frequency motions.
-F Define a force field to build your systems. <OPLS_2005>
OPLS_2005, S-OPLS, OPLS3e, OPLS3, OPLS2 are recommended to protein-ligand systems.
Simulation control parameter:
-m Enter the maximum simulation time for the Brownian motion simulation, in ps. <100>
-t Enter the Molecular dynamics simulation time for the product simulation, in ns. <100>
-T Specify the temperature to be used, in kelvin. <310>
-N Number of Repeat simulation with different random numbers. <1>
-P Define a ASL to protein, such as "protein".
-L Define a ASL to ligand, such as "res.ptype UNK".
-q Turn off protein-ligand analysis.
-u Turn off md simulation, only system build.
-C Set constraint to an ASL, such as "chain.name A AND backbone"
-f Set constraint force, default is 10.
-o Specify the approximate number of frames in the trajectory. <1000>
This value is coupled with the recording interval for the trajectory and the simulation time: the number of frames times the trajectory recording interval is the total simulation time.
If you adjust the number of frames, the recording interval will be modified.
Job control:
-G HOST of GPU queue, default is HPC_GPU.
-H HOST of CPU queue, default is HPC_CPU.
-D Your Desmond path. <$Desmond>2、AutoMD: Desmond分子动力学模拟一键式运行脚本:
下载地址:Wang-Lin-boop/AutoMD: Easy to get started with molecular dynamics simulation. (github.com)
脚本介绍:AutoMD:从初始结构到MD轨迹,只要一行Linux命令? - 知乎 (zhihu.com)
由于Desmond学术版本提供的力场有限,plmd无法有效引入其他力场参数,因此在plmd的迭代版本AutoMD上,通过配置D.E.Shaw Research开发的Viparr和Msys代码在Desmond中引入如Amber,Charmm等力场来帮助模拟系统的构建。
在个人笔记本(Linux)上下载以下代码文件,并上传至集群个人home的software中:
wget https://github.com/DEShawResearch/viparr/releases/download/4.7.35/viparr-4.7.35-cp38-cp38-manylinux2014_x86_64.whl
wget https://github.com/DEShawResearch/msys/releases/download/1.7.337/msys-1.7.337-cp38-cp38-manylinux2014_x86_64.whl
git clone git://github.com/DEShawResearch/viparr-ffpublic.git
git clone https://github.com/Wang-Lin-boop/AutoMD随后进入desmond工作目录,启动虚拟环境以帮助安装Viparr和Msys:
cd /fsa/home/ljx_zhangzw/software
./run schrodinger_virtualenv.py schrodinger.ve
source schrodinger.ve/bin/activate
pip install --upgrade pip
pip install msys-1.7.337-cp38-cp38-manylinux2014_x86_64.whl
pip install viparr-4.7.35-cp38-cp38-manylinux2014_x86_64.whl
echo "export viparr=${PWD}/schrodinger.ve/bin" >> ~/.bashrc同时,将viparr-ffpublic.git解压并添加到环境变量:
echo "export VIPARR_FFPATH=${PWD}/viparr-ffpublic/ff" >> ~/.bashrc最后,将AutoMD.git解压并进行安装,同时指定可自动添加补充力场:
cd AutoMD
echo "alias AutoMD=${PWD}/AutoMD" >> ~/.bashrc
chmod +x AutoMD
source ~/.bashrc
cp -r ff/* ${VIPARR_FFPATH}/#AutoMD -h
在输入命令介绍中,给出了四个示例:胞质蛋白-配体复合物、血浆蛋白-蛋白复合物、DNA/RNA-蛋白质复合物、以及需要Meastro预准备的膜蛋白。在这些示例中:
通过-i命令输入了当前路径所有的复合物结构.mae文件(也可以是Maestro构建好的模拟系统.cms文件);
通过-S指定了模拟系统的溶液环境,包括INC(0.15M KCl、SPC水、K+中和)、OUC(0.15M NaCl、SPC水、Na+中和),以及自定义溶液环境如"SPC:Cl:0.15-K-Cl+0.02-Mg2-Cl";
通过-b和-s定义了模拟系统的Box形状和大小;
通过-F定义力场参数,由于学术版Desmond的要求不被允许使用S-OPLS力场,仅可使用OPLS_2005或其他力场;OPLS适用于蛋白-配体体系,Amber适用于蛋白-核酸,Charmm适用于膜蛋白体系,DES-Amber适用于PPI复合体体系,但是这些搭配并不是绝对的,我们当然也可以尝试在膜蛋白体系上使用Amber力场,在核酸体系上使用Charmm力场,具体的情况需要用户在自己的体系上进行尝试;
| -F Charmm ff | aa.charmm.c36m, misc.charmm.all36, carb.charmm.c36, ethers.charmm.c35, ions.charmm36, lipid.charmm.c36 and na.charmm.c36 |
| -F Amber ff | aa.amber.19SBmisc, aa.amber.ffncaa, lipid.amber.lipid17, ions.amber1lm_iod.all, ions.amber2ff99.tip3p, na.amber.bsc1 and na.amber.tan2018 |
| -F DES-Amber | aa.DES-Amber_pe3.2, dna.DES-Amber_pe3.2, rna.DES-Amber_pe3.2 and other force fields in -F Amber |
Usage: AutoMD [OPTION] <parameter>
Example:
1) MD for cytoplasmic protein-ligand complex:
AutoMD -i "*.mae" -S INC -P "chain.name A" -L "res.ptype UNK" -F "S-OPLS"
2) MD for plasma protein-protein complex:
AutoMD -i "*.mae" -S OUC -F "DES-Amber"
3) MD for DNA/RNA-protein complex:
AutoMD -i "*.mae" -S "SPC:Cl:0.15-K-Cl+0.02-Mg2-Cl" -F Amber
4) MD for membrane protein, need to prior place membrane in Meastro.
AutoMD -i "*.mae" -S OUC -l "POPC" -r "Membrane" -F "Charmm"
Input parameter:
-i Use a file name (Multiple files are wrapped in "", and split by ' ') *.mae or *.cms ;
or regular expression to represent your input file, default is *.mae.
System Builder parameter:
-S System Build Mode: <INC>
INC: System in cell, salt buffer is 0.15M KCl, water is SPC. Add K to neutralize system.
OUC: System out of cell, salt buffer is 0.15M NaCl, water is SPC. Add Na to neutralize system.
Custom Instruct: Such as: "TIP4P:Cl:0.15-Na-Cl+0.02-Fe2-Cl+0.02-Mg2-Cl"
Interactive addition of salt. Add Cl to neutralize system.
for positive_ion: Na, Li, K, Rb, Cs, Fe2, Fe3, Mg2, Ca2, Zn2 are predefined.
for nagative_ion: F, Cl, Br, I are predefined.
for water: SPC, TIP3P, TIP4P, TIP5P, DMSO, METHANOL are predefined.
-l Lipid type for membrane box. Use this option will build membrane box. <None>
Lipid types: POPC, POPE, DPPC, DMPC.
-b Define a boxshape for your systems. <cubic>
box types: dodecahedron_hexagon, cubic, orthorhombic, triclinic
-s Define a boxsize for your systems. <15.0>
for dodecahedron_hexagon and cubic, defulat is 15.0;
for orthorhombic or triclinic box, defulat is [15.0 15.0 15.0];
If you want use Orthorhombic or Triclinic box, your parameter should be like "15.0 15.0 15.0"
-R Redistribute the mass of heavy atoms to bonded hydrogen atoms to slow-down high frequency motions.
-F Define a force field to build your systems. <OPLS_2005>
OPLS_2005, S-OPLS are recommended to receptor-ligand systems.
Amber, Charmm, DES-Amber are recommended to other systems. Use -O to show more details.
Use the "Custom" to load parameters from input .cms file.
Simulation control parameter:
-m Enter the maximum simulation time for the Brownian motion simulation, in ps. <100>
-r The relaxation protocol before MD, "Membrane" or "Solute". <Solute>
-e The ensemble class in MD stage, "NPT", "NVT", "NPgT". <NPT>
-t Enter the Molecular dynamics simulation time for the product simulation, in ns. <100>
-T Specify the temperature to be used, in kelvin. <310>
-N Number of Repeat simulation with different random numbers. <1>
-P Define a ASL to receptor, such as "protein".
-L Define a ASL to ligand and run interaction analysis, such as "res.ptype UNK".
-u Turn off md simulation, only system build.
-C Set constraint to an ASL, such as "chain.name A AND backbone"
-f Set constraint force, default is 10.
-o Specify the approximate number of frames in the trajectory. <1000>
This value is coupled with the recording interval for the trajectory and the simulation time:
the number of frames times the trajectory recording interval is the total simulation time.
If you adjust the number of frames, the recording interval will be modified.
Job control:
-G HOST of GPU queue, default is GPU.
-H HOST of CPU queue, default is CPU.
-D Your Desmond path. </fsa/home/ljx_zhangzw/>
-V Your viparr path. </fsa/home/ljx_zhangzw/schrodinger.ve/bin>
-v Your viparr force fields path. </fsa/home/ljx_zhangzw/viparr-ffpublic/ff>示例任务提交脚本及命令,需修改相应计算节点和软件路径信息:
#BSUB -L /bin/bash
#BSUB -q 83a100ib
#BSUB -m m001
module load cuda/12.0.0
export Desmond=/fsa/home/ljx_zhangzw/
export viparr=/fsa/home/ljx_zhangzw/schrodinger.ve/bin
export VIPARR_FFPATH=/fsa/home/ljx_zhangzw/viparr-ffpublic/ff
alias AutoMD=/fsa/home/ljx_zhangzw/AutoMD/AutoMD
echo $PATH
echo $LD_LIBRARY_PATH
nvidia-smi
env|grep CUDA
AutoMD -i "*.mae" -S OUC -P "chain.name A" -L "res.ptype UNK" -F "OPLS_2005" -T 298 -s 10 -H m001 -G m001在模拟参数控制命令中,通过-P和-L分别定义了mae文件中的受体和配体,-m默认
-T定义体系温度为298 K,默认进行100 ns和1次重复模拟次数,通过-H和-G指定了host文件中的相应计算队列。
四、性能表现:
笔者通过上述的示例任务提交脚本及命令调用Desmond和AutoMD进行了包含12个阶段的工作流:
Summary of user stages:
stage 1 - task
stage 2 - build_geometry
stage 3 - assign_forcefield
stage 4 - simulate, Brownian Dynamics NVT, T = 10 K, small timesteps, and restraints on solute_heavy_atom, 100 ps
stage 5 - simulate, Brownian Dynamics NVT, T = 10 K, small timesteps, and restraints on user defined sets, 100 ps
stage 6 - simulate, Langevin small steps NVT, T = 10 K, and restraints on solute heavy atoms, 12 ps
stage 7 - simulate, Langevin NPT, T = 10 K, and restraints on solute heavy atoms, 12 ps
stage 8 - simulate, Langevin NPT, T = 10 K, and restraints on solute heavy atoms, 12 ps
stage 9 - simulate, Langevin NPT and restraints on solute heavy atoms, 12ps
stage 10 - simulate, Langevin NPT and no restraints, 24ps
stage 11 - simulate, Final MD and analysis, 100000.0 ps
stage 12 - pl_analysis
(12 stages in total)分别在83a100ib和72rtxib进行了相应尝试:
System Atoms:21173
System Residues:6363
System MOls:6202
Protein Atoms:2584
Protein Residues:162
Ligand Atoms:73
Ligand Residues:1
MIN time: 100 ps
MD time: 100000.0 ps
temperature: 298 K
Repeat: 1
Rondom numbers list: 2007
#Desmond
Multisim summary :
Total duration: 3h 38' 7"
Total GPU time: 3h 24' 42" (used by 8 subjob(s))
#HPC-83a100ib
Resource usage summary:
CPU time : 12745.00 sec.
Max Memory : 965 MB
Average Memory : 893.78 MB
Total Requested Memory : -
Delta Memory : -
Max Swap : -
Max Processes : 15
Max Threads : 45
Run time : 13262 sec.
Turnaround time : 13260 sec.System Atoms:63284
System Residues:20178
System MOls:19955
Protein Atoms:2585
Protein Residues:224
Ligand Atoms:72
Ligand Residues:1
MIN time: 100 ps
MD time: 100000.0 ps
temperature: 298 K
Repeat: 1
Rondom numbers list: 2007
#Desmond
Multisim summary :
Total duration: 7h 51' 5"
Total GPU time: 7h 27' 48" (used by 8 subjob(s))
#HPC-72rtxib
Resource usage summary:
CPU time : 31316.00 sec.
Max Memory : 1634 MB
Average Memory : 400.16 MB
Total Requested Memory : -
Delta Memory : -
Max Swap : -
Max Processes : 15
Max Threads : 40
Run time : 85601 sec.
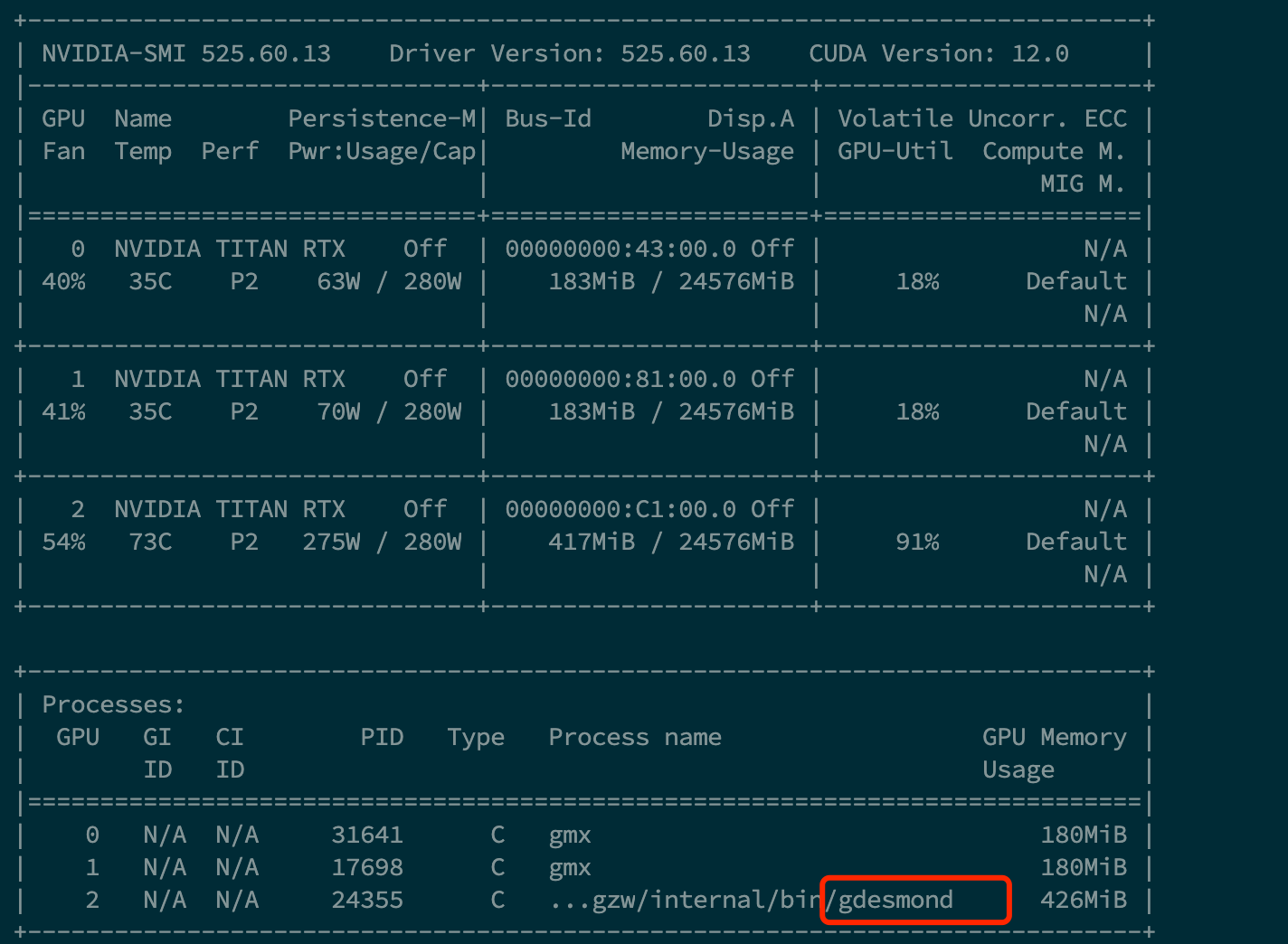
Turnaround time : 85602 sec.笔者在测试时恰逢72rtxib g007中有同学在运行gmx,desmond的GPU利用率得到了进一步的凸显,但目前存在一点问题,即desmond在计算完毕后无法及时退出对GPU的占用,
***我将在错误解决后更新,或者如果你知道如何排除这个出错误,请在QQ群探讨***
五、模拟分析:
模拟完成后,将在工作路径创建一个名为结构文件名_随机种子号-md的文件夹,其中包含了:
| 多个阶段的工作流文件 | AutoMD.msj |
| 多个阶段 launching日志文件 | md_multisim.log |
| 多个阶段 launching阶段文件 | md_1-out.tgz ... md_12-out.tgz |
| 模拟系统结构文件 | -in.cms and -out.cms |
| 模拟执行参数文件 | md.cpt.cfg and md-out.cfg |
| 模拟轨迹文件夹 | md_trj |
| 模拟执行日志文件 | md.log |
| 检查点文件 | md.cpt |
| 能量文件 | md.ene |
| 包含RMSF、RMSF、SSE等常规分析的结果文件 | md.eaf |
此外,还包含了部分的分析脚本文件,更多用于MD轨迹分析的脚本文件还可以通过访问官方脚本库获得Scripts | Schrödinger (schrodinger.com);
| Resume previous MD | bash resume.sh |
| Extend MD | bash extend.sh # The total time will be extend to 2-fold of initial time |
| Cluster trajectory | bash cluster.sh "<rmsd selection>" "<fit selection>" "<number>" |
| Analysis Occupancy | bash occupancy.sh "<selection to analysis>" "<fit selection>" |
| Analysis ppi contact | bash ppi.sh "<Component A>" "<Component B>" |
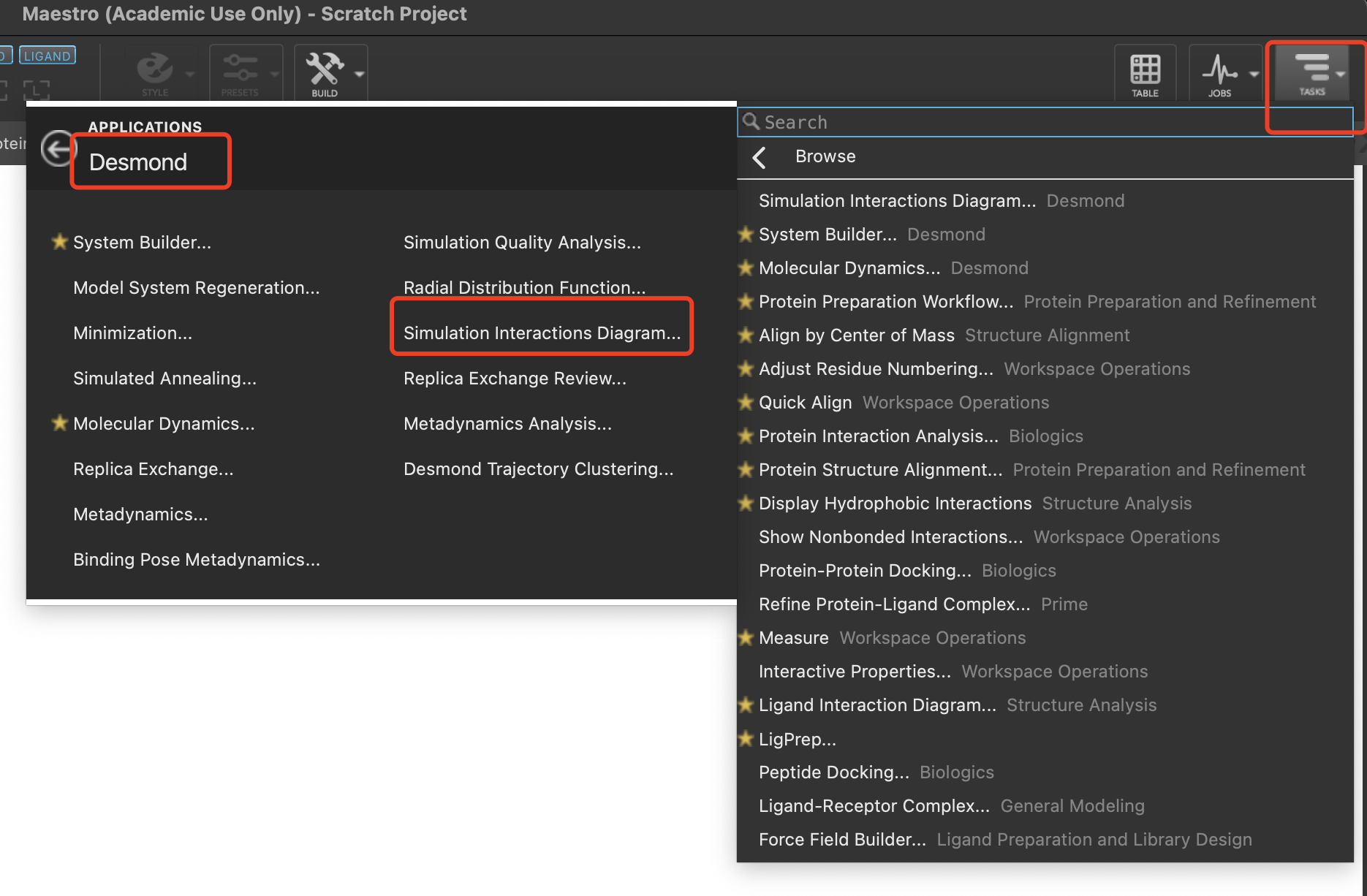
下载结构文件名_随机种子号-md的文件夹至个人笔记本,在TASKS中的Desmond找到Simulation Interaction Digram分析面板,并导入md.eaf文件进行常规分析。
六、致谢:
需要强调的是,AutoMD是一个建立在Desmond软件包基础上的脚本,模拟的质量由Desmond本身决定,脚本没有对模拟程序本身做任何的改动,且所有的学术贡献归功于D. E. Shaw Research (无商业利益相关)。
AutoMD作为一个使用脚本,仅仅是为了解决了一些实际MD应用中的问题,方便初学者通过这个脚本快速上手MD,但请勿认为AutoMD是一种MD模拟的程序,其内核仍然是Desmond程序。
本文所有内容均来自上海科技大学Wang-Lin,笔者整理使用经验而已。